简介
如图所示,在AspenV15版本中,新增了一个Mol-Instincts数据库,也就是顶部的ChemEssen化工按钮:
Mol-Instincts数据库 是一个专注于化学与材料科学 的综合性数据平台,主要用于提供分子、材料及相关属性的详细信息和计算工具。
主要功能与数据内容:
- 分子/材料数据库:包含大量化合物(有机、无机、聚合物等)的物理化学性质(如熔点、沸点、溶解度)、光谱数据(IR、NMR)、热力学参数等。
- 预测工具:基于算法和模型预测分子性质(如毒性、反应活性)、材料性能(如导电性、机械强度)等。
- 反应路径分析:部分版本可能提供化学反应机理或合成路径的模拟数据。
- 3D结构数据:提供分子的三维构象优化结构,支持可视化与分析。
核心建模方法:
(1) 量子化学计算
- 密度泛函理论(DFT):用于预测分子电子结构、热力学性质(如生成焓)、光谱数据(IR/NMR)等。
- 分子动力学(MD):模拟材料动态行为(如扩散系数、机械性能)。
- 半经验方法(如PM6、AM1):快速估算大分子体系的能量和构型。
(2) 机器学习(ML)与AI模型
- 描述符(Descriptor):基于分子指纹(如Morgan指纹)、拓扑或几何特征(如QM9数据集)构建模型。
- 常见算法:
- 传统ML:随机森林(RF)、支持向量机(SVM)用于分类/回归任务(如毒性预测)。
- 深度学习:图神经网络(GNN)、Transformer模型(如用于反应预测或性质优化)。
- 预训练模型:可能利用类似ChemBERTa或MGNN(分子图神经网络)的迁移学习技术。
(3) 混合方法(Hybrid)
- QM/ML组合:量子力学计算提供高精度数据,ML模型加速预测(如用DFT数据训练GNN预测能垒)。
- 多任务学习:同时预测多个性质(如溶解度+LogP),提升泛化性。
官网: ChemEssen l Total Solution for the Chemical and Drone Industries
IK-CAPE文件:
在 80 年代后期,一个名为 IK-CAPE 的德国公司联盟开发了一种例程规范,用于计算 FORTRAN 中的热物理特性。此外,还开发了基于该规范的实现方案,并将其用于多个内部仿真代码和学术研究项目。在与拜耳技术服务公司的联合项目中,AixCAPE 为该 IK-CAPE 软件包配备了 CAPE-OPEN 热力学接口,以允许在最先进的仿真环境中使用现有的属性数据输入文件。使用 Aspen Plus、PRO/II、COFE、gPROMS 和 Simulis Thermodynamics 进行了成功的测试。
正文
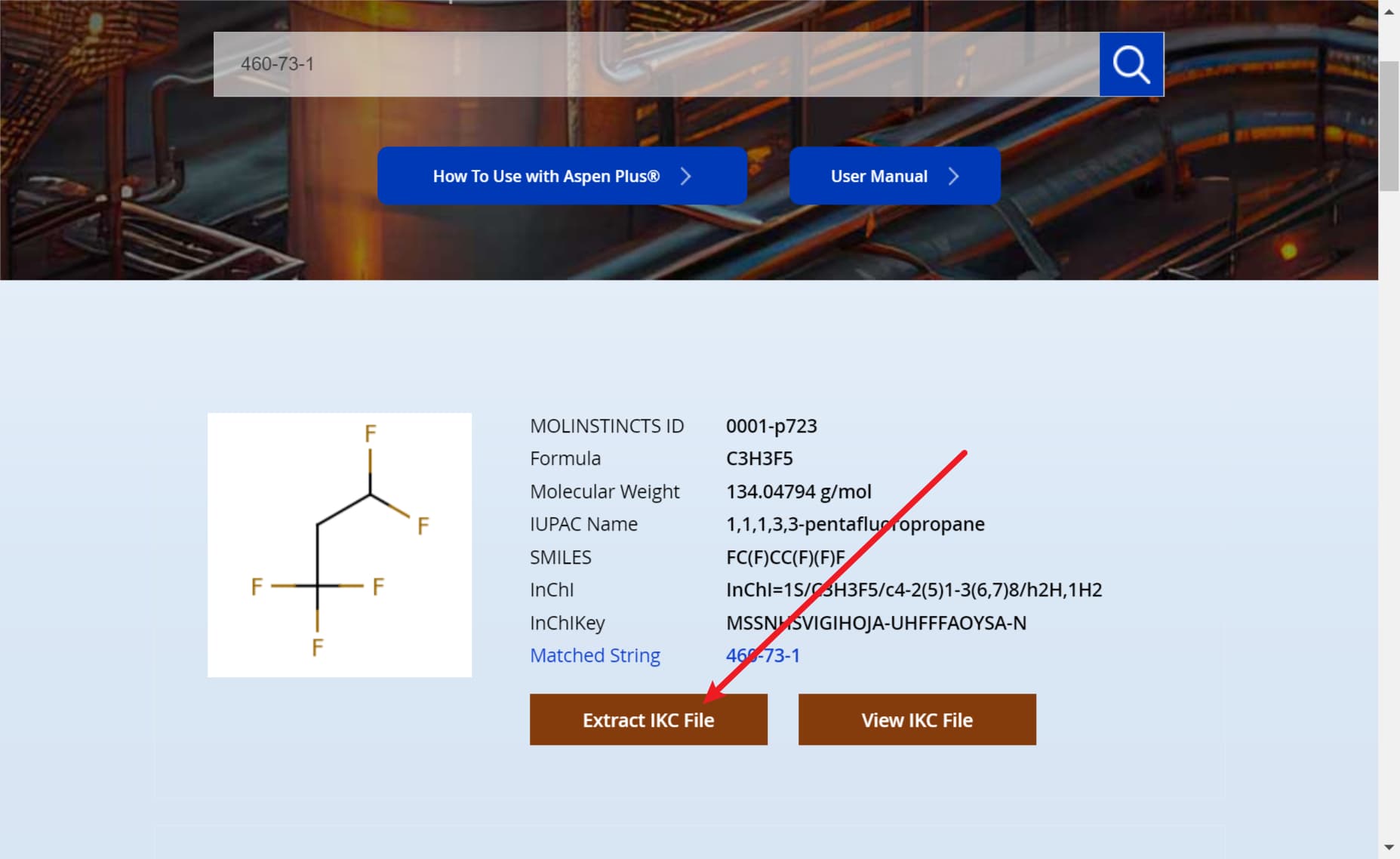
本文以1,1,1,3,3-五氟丙烷(460-73-1)和1,1,1,2-四氟乙烷(811-97-2)为例,首先可以看到数据库里是有的,但是没有二元交互参数:
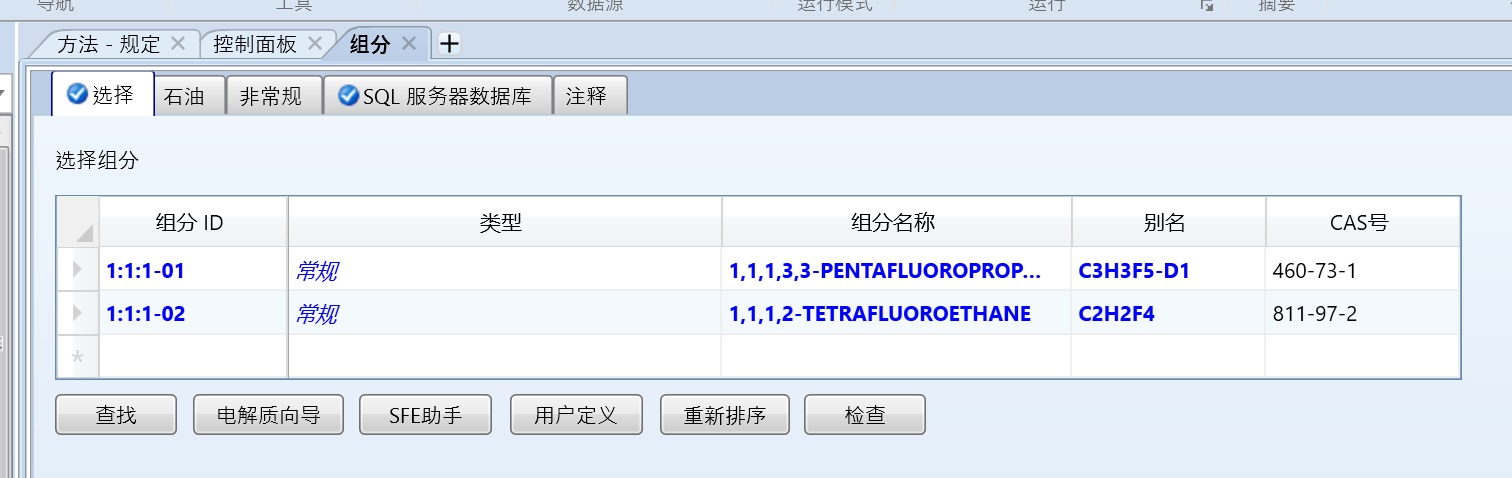
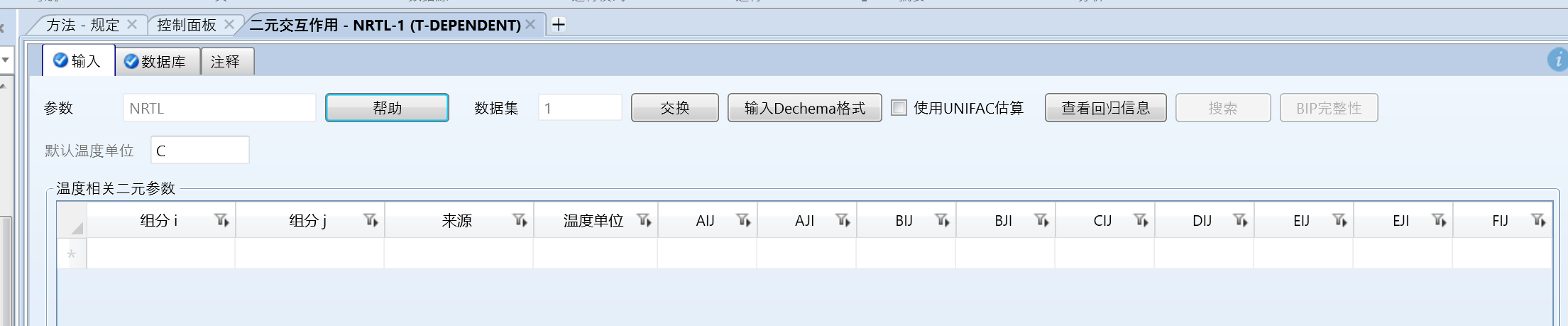
我们示例中不使用自带数据库的组分数据,这里只是演示一下没有交互参数。
导入文件
新建一个模拟,然后点击顶部的ChemEssen化工按钮,点击确定,出现导入文件选项:
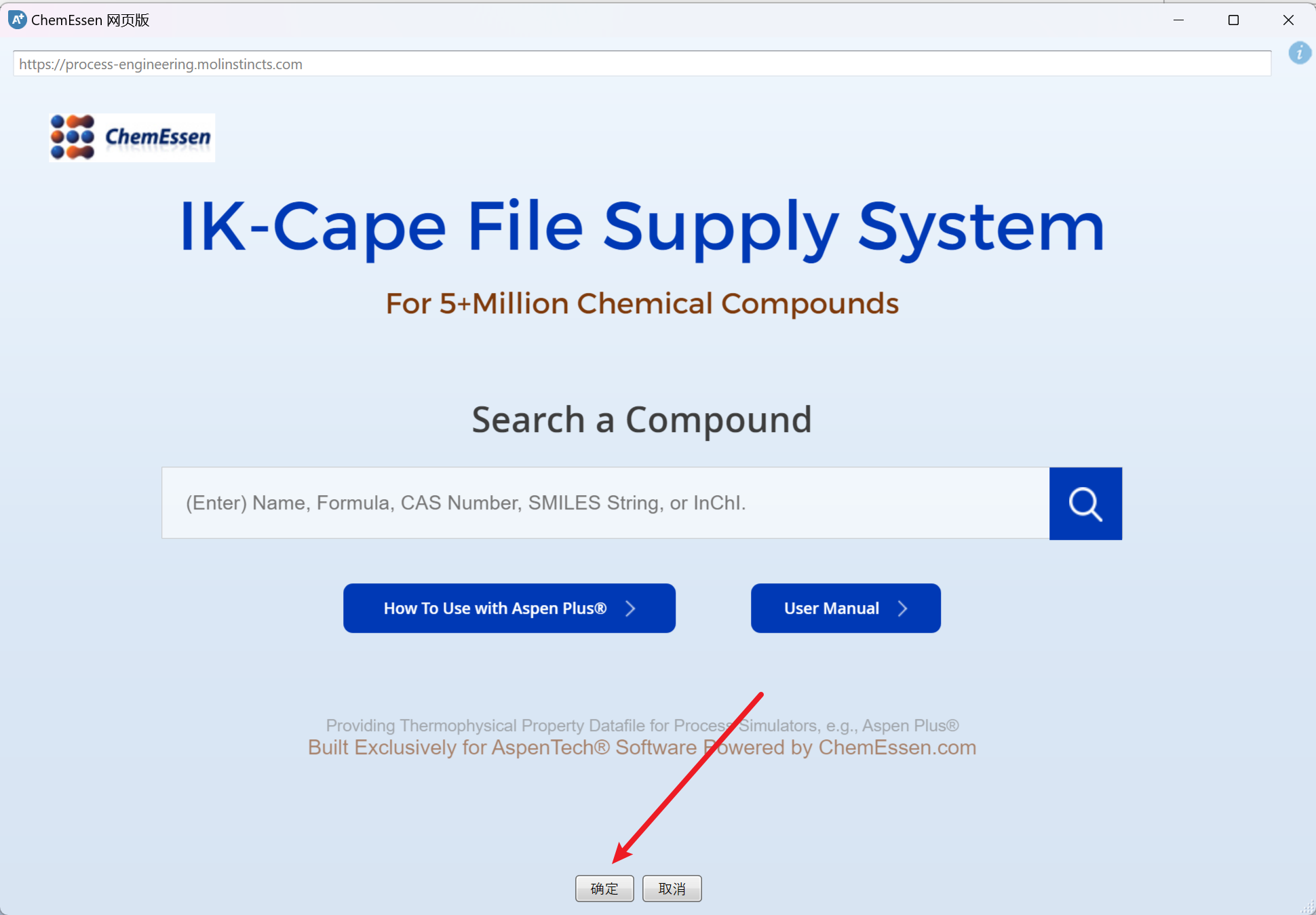
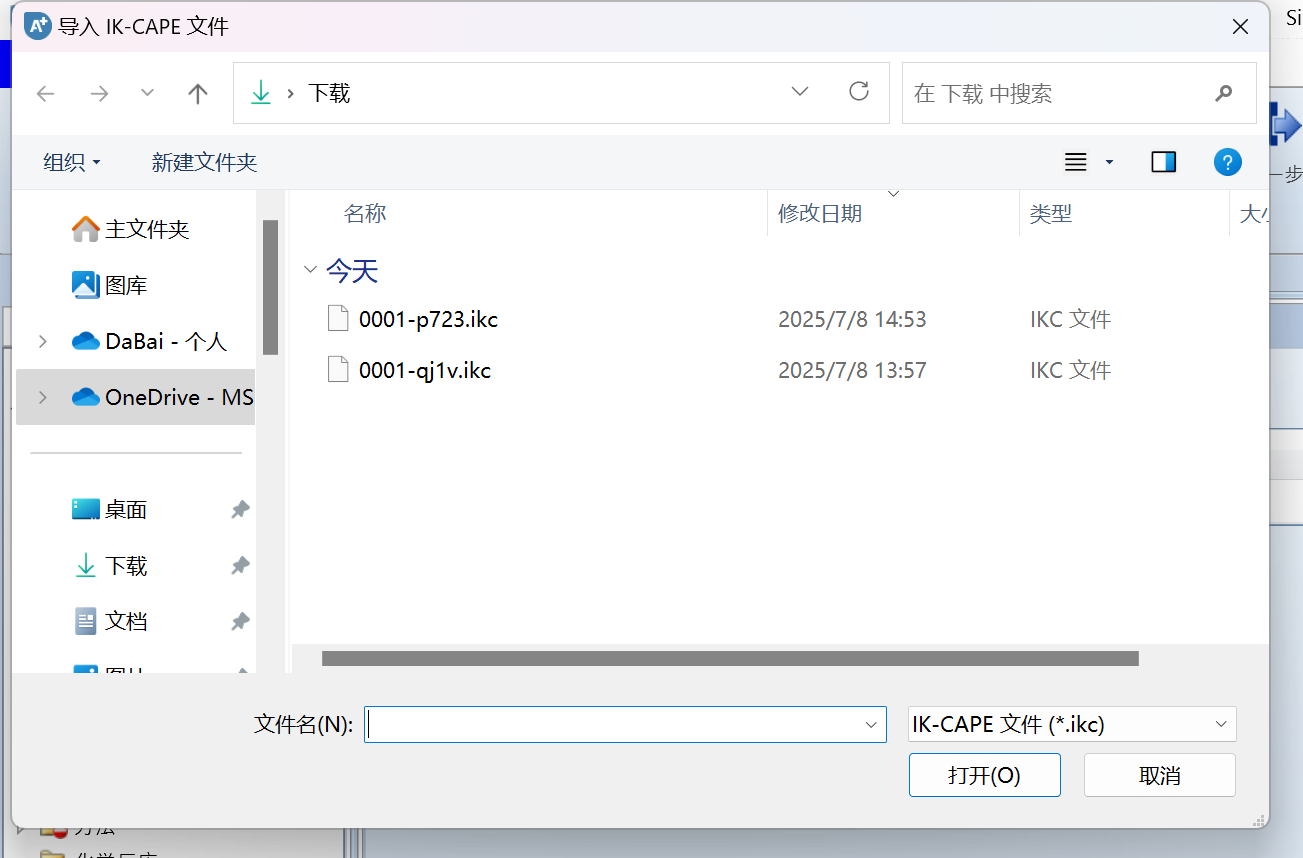
选择要添加的ikc文件,点击打开,即可导入,如果出现需要解决ID冲突(也就是数据库中有该组分),先点击合并,再点击确定:
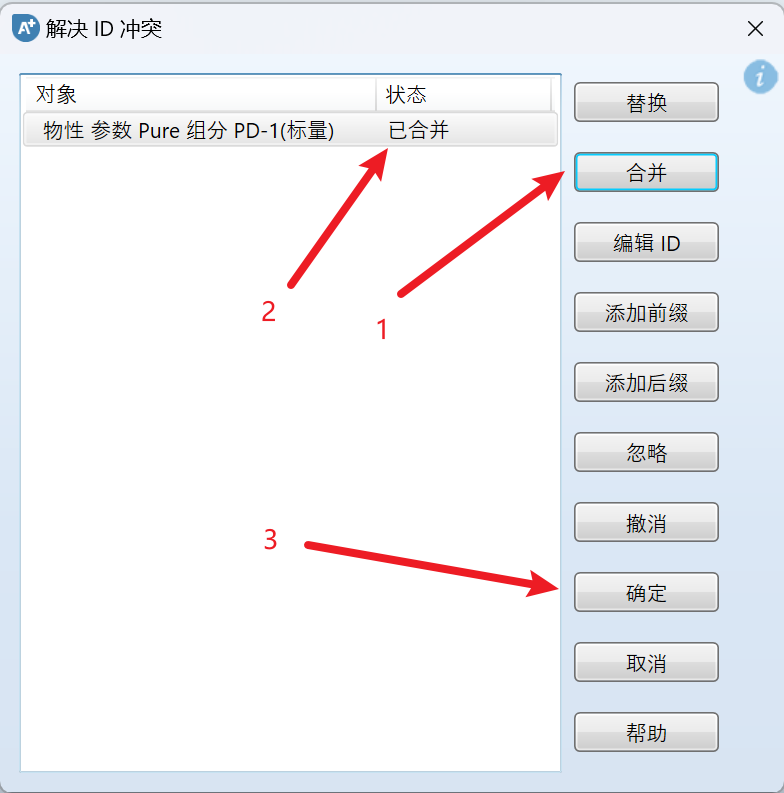
然后点击确定插入:
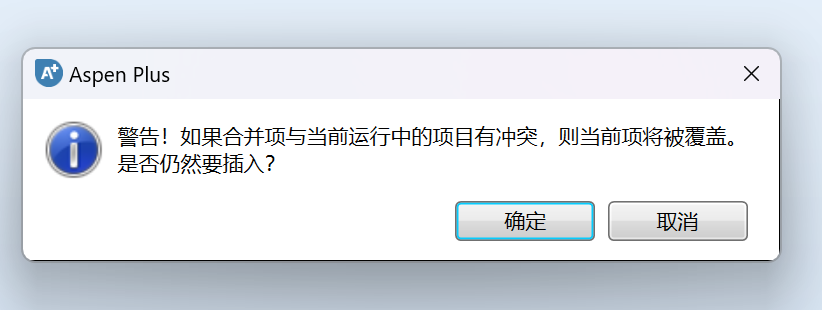
导入结束:
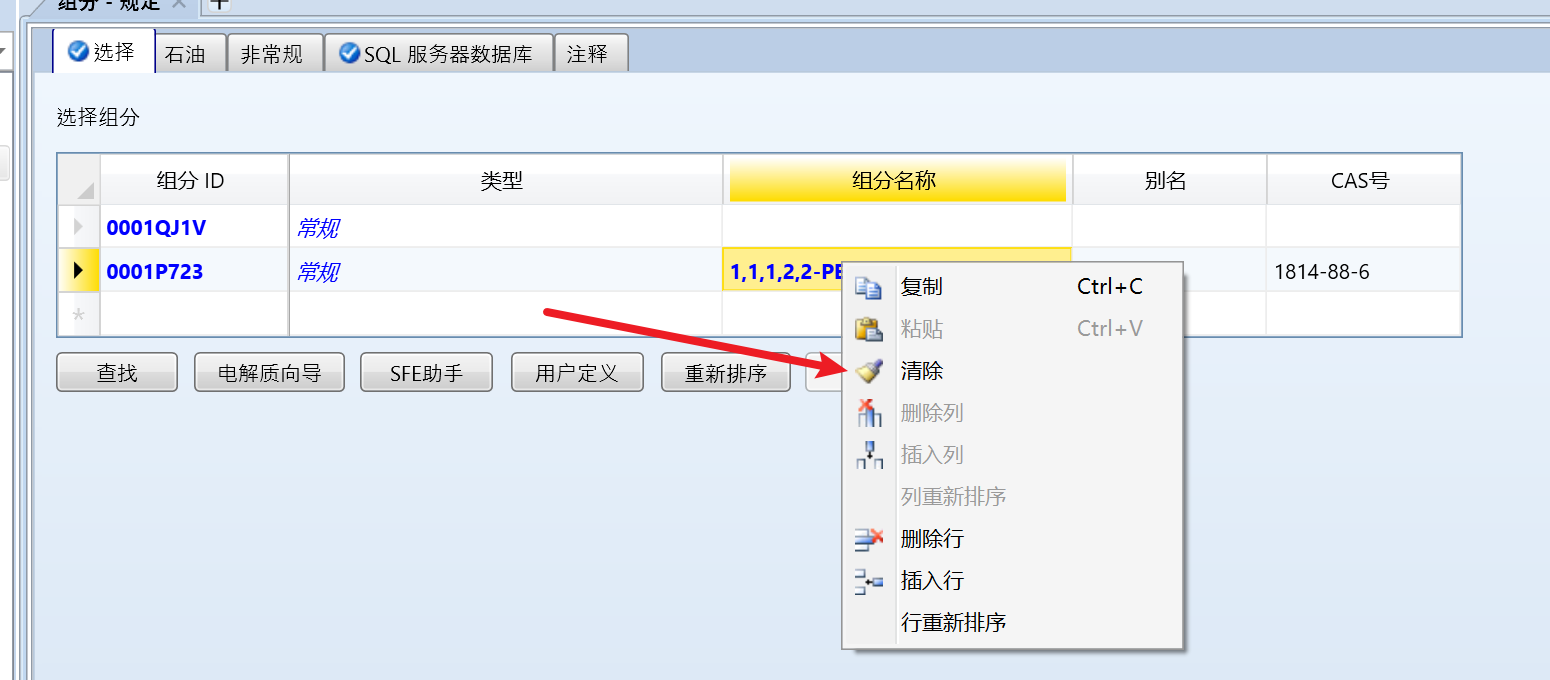
后可以右键组分名称,点击清除:
验证文件
点击左侧的方法、参数、纯组分、PD-1:
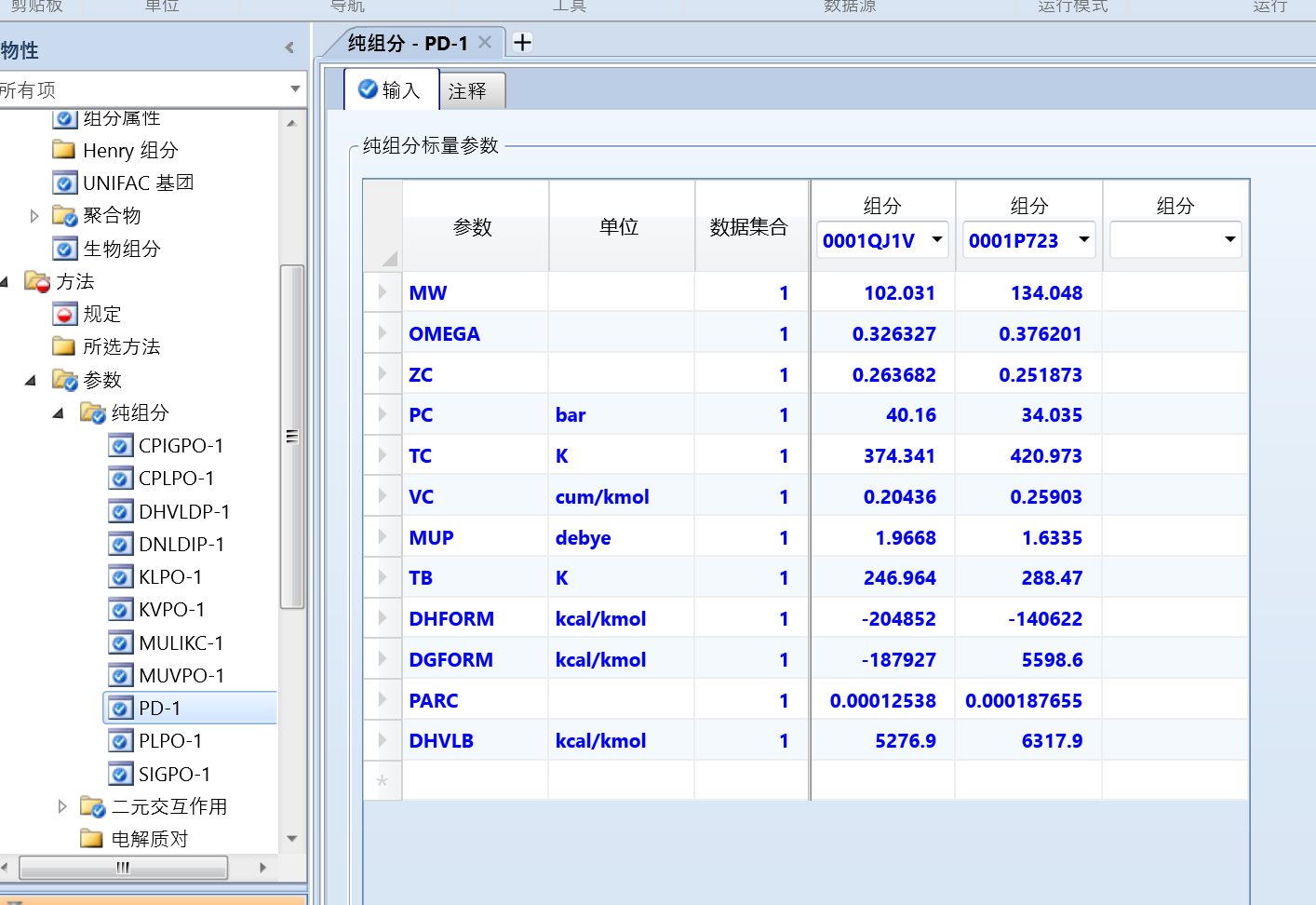
可以看到导入进来的很多参数。
绘制结构
点击组分、分子结构,点击结构和官能团、绘图/导入/编辑:
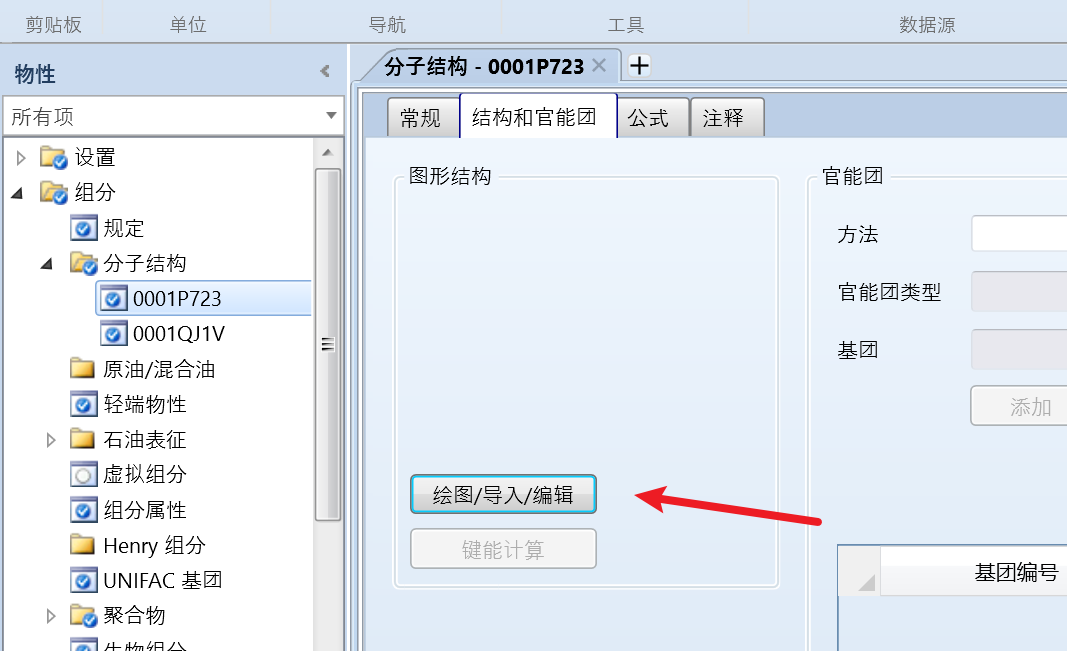
绘制或者导入对应组分的结构:
这里非常不建议自己画结构,因为可能自己画的结构会导致键能计算错误而无法估算,强烈建议直接使用 SMILES 格式直接导入,也就是
*.smi文件。
SMILES查询推荐可以使用免费开源的PubChem数据库: PubChem
这里的 SMILES 格式是什么,我之前有文章提及过,有兴趣可以回顾一下:
分子结构中的SMILES - 化学工程 - CEPD@BBS
使用ChemDraw输入3D分子结构到COSMOThermX中 - 化学工程 - CEPD@BBS
点击左上角的文件夹图标,选择 *.smi 文件格式,导入:
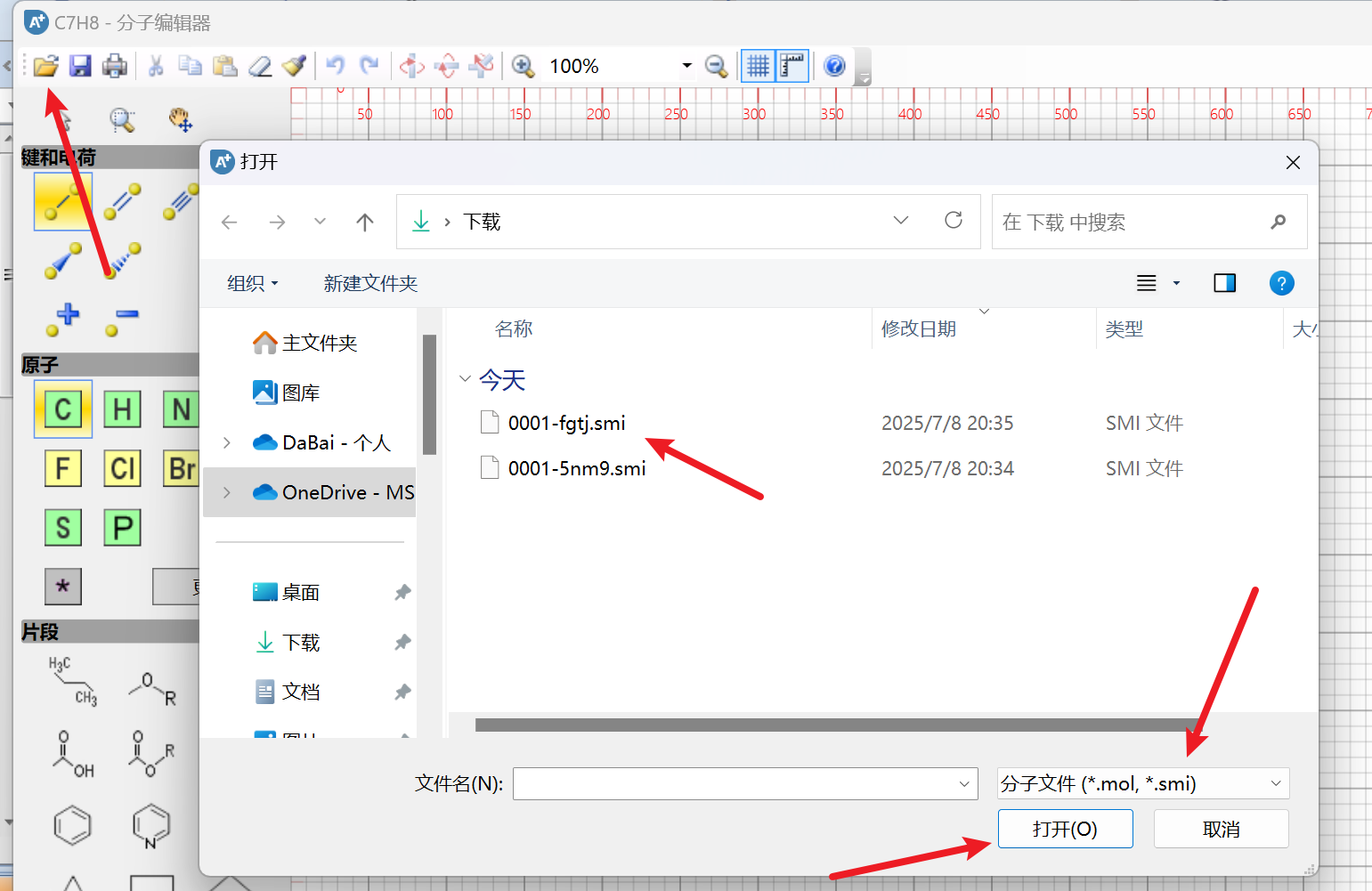
绘制好或者导入后,点击右上角的x即可保存:
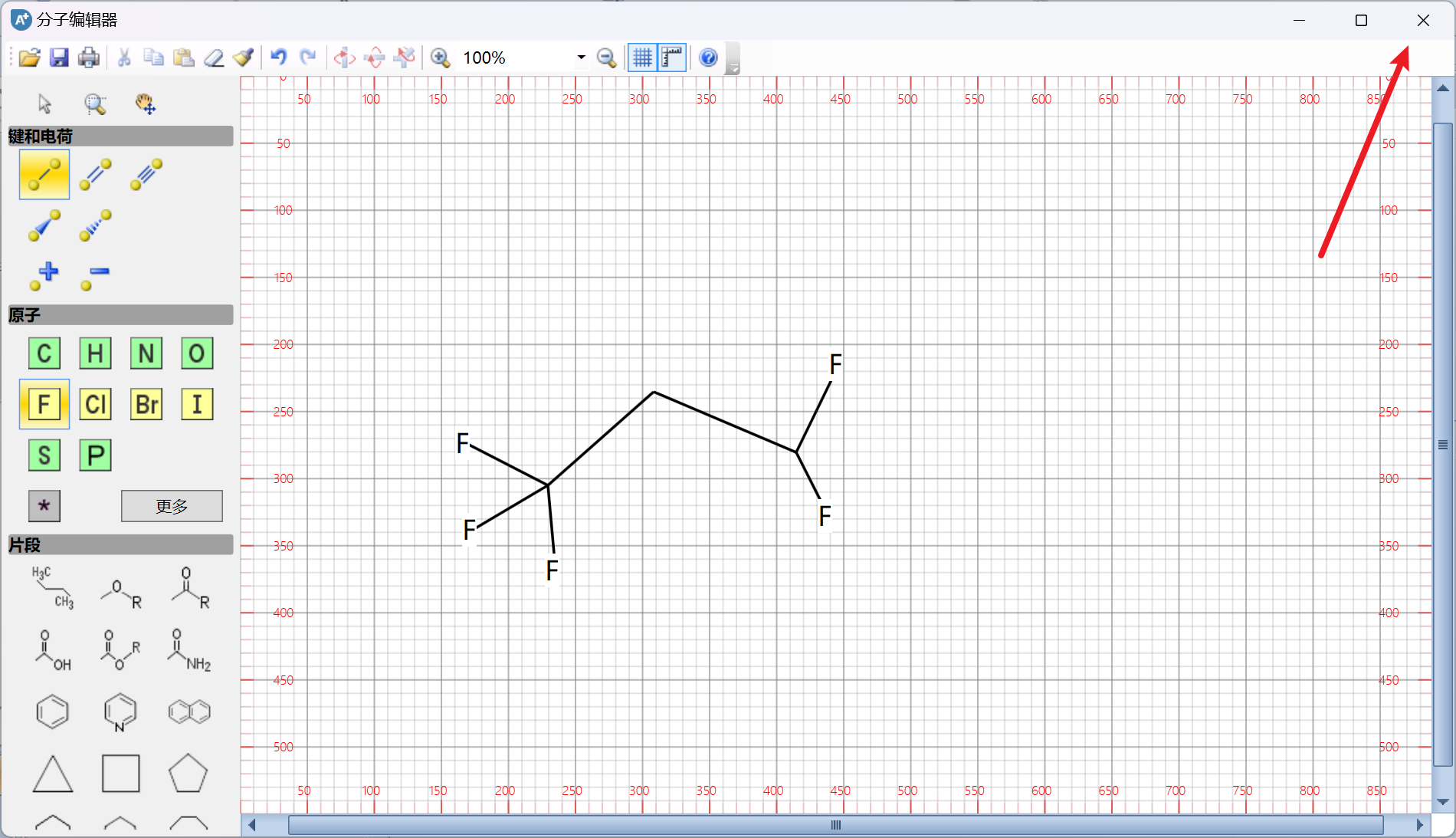
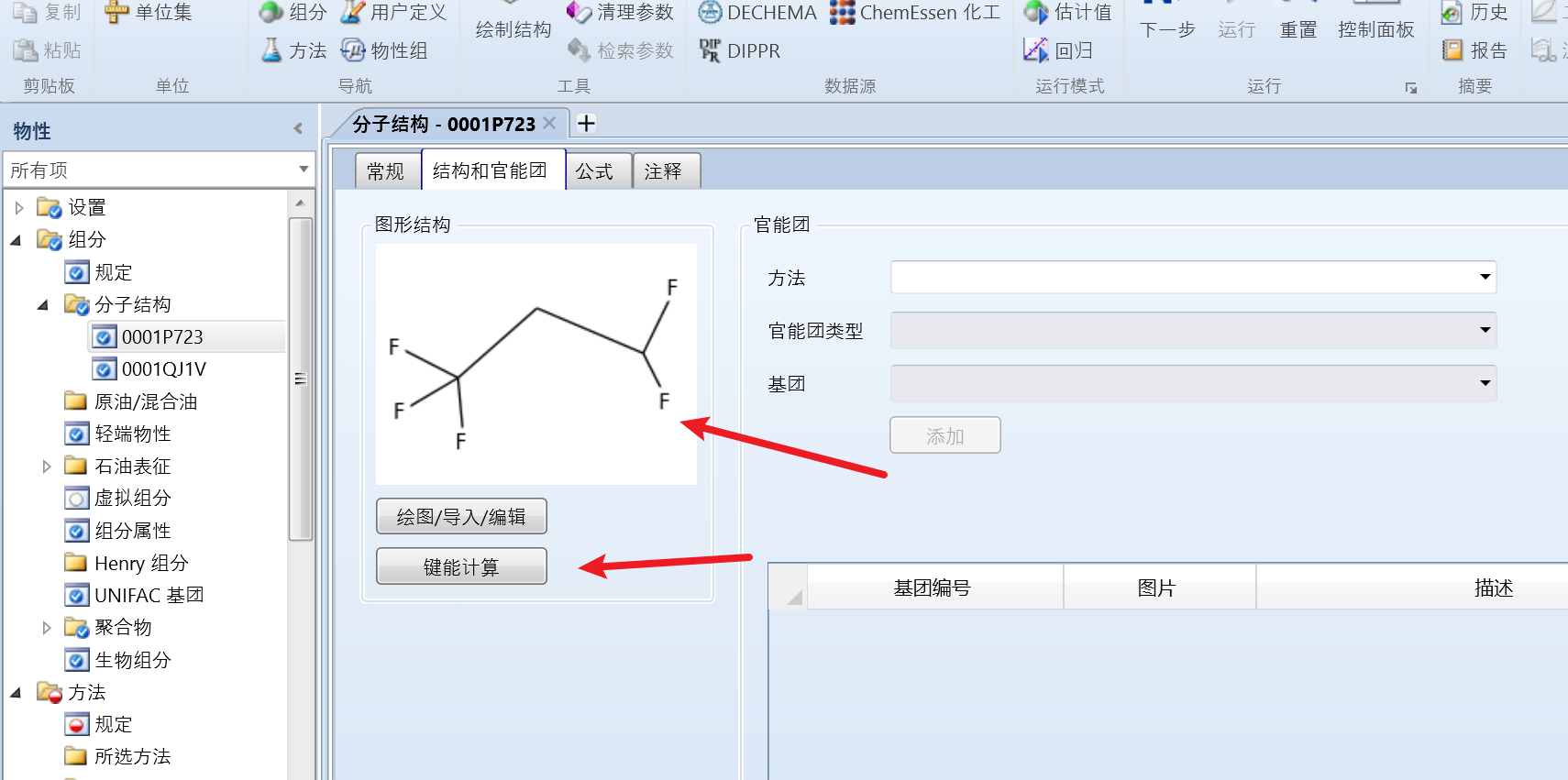
然后点击键能计算,即可自动识别分子键和公式:
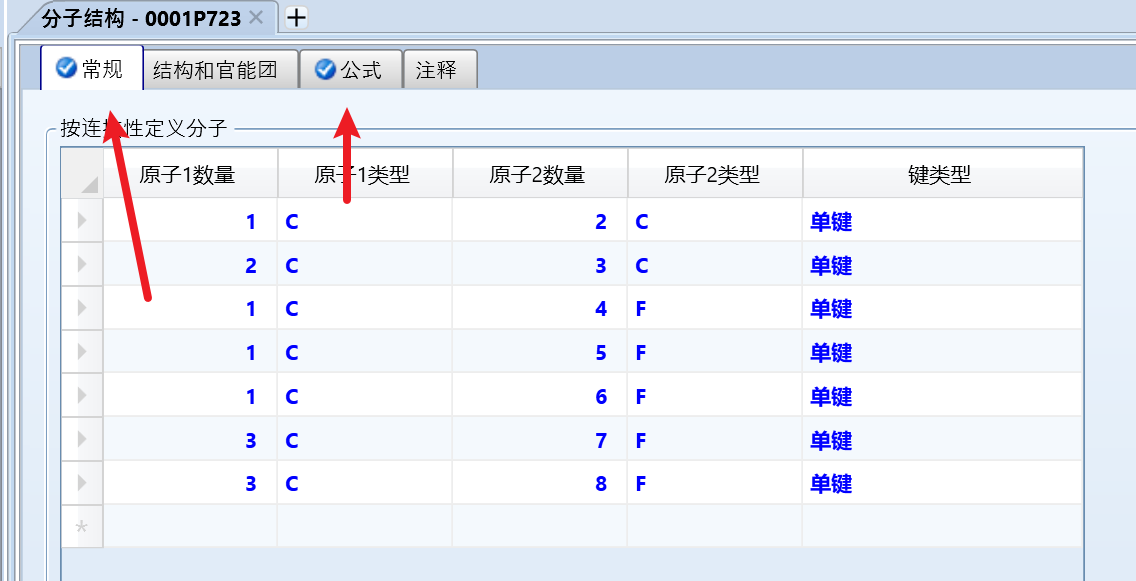
另一个组分同样的操作即可。
估算二元参数
设置好物性方法(以NRTL为例):
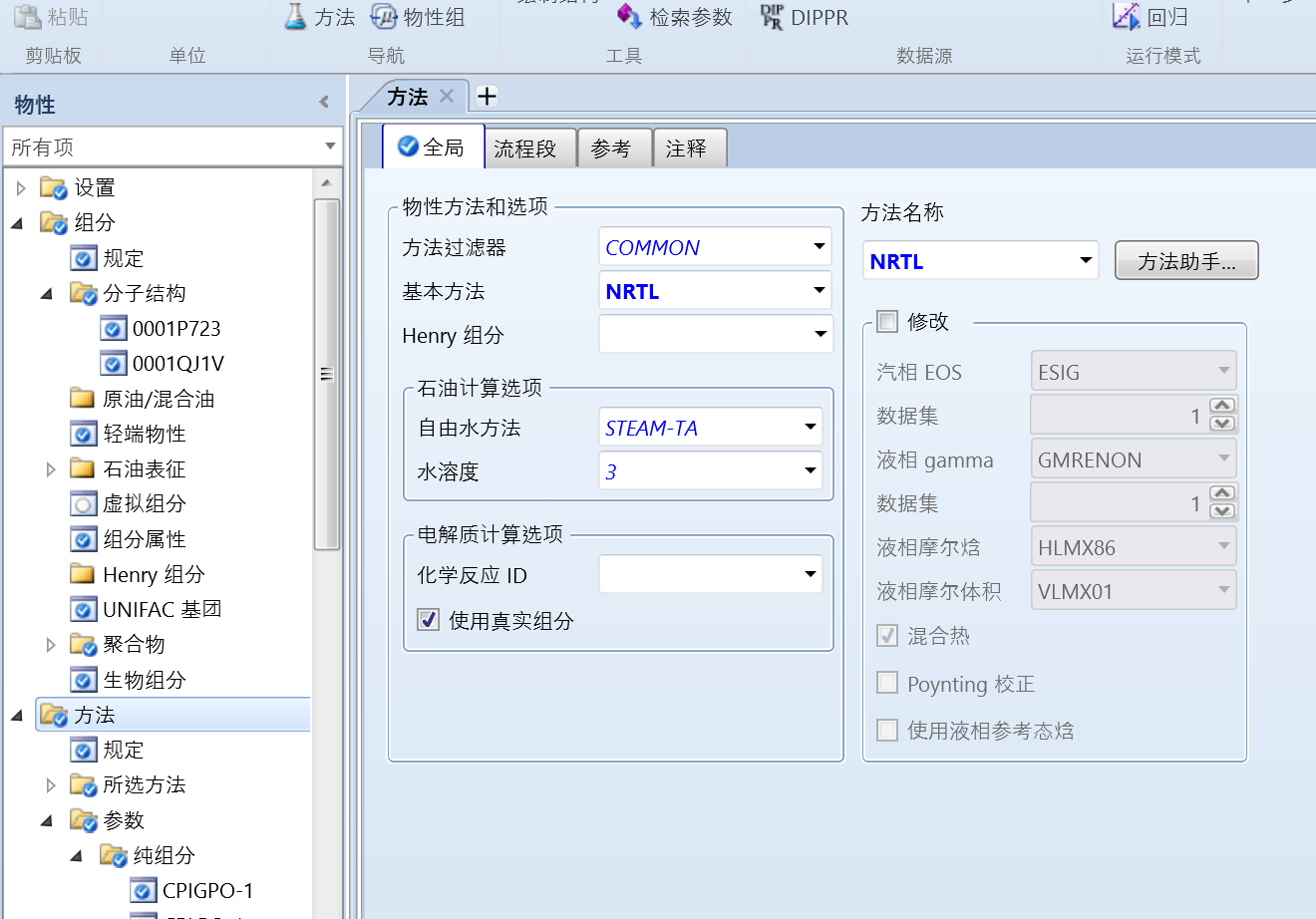
确认二元参数为空:
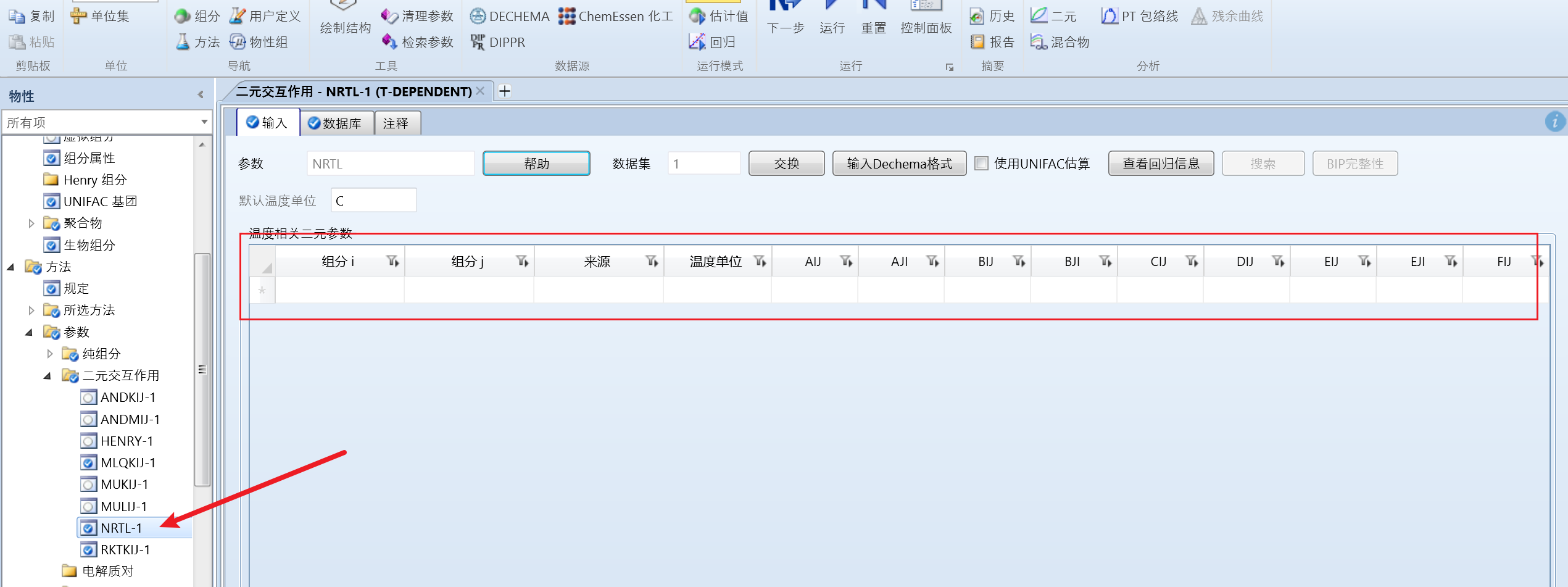
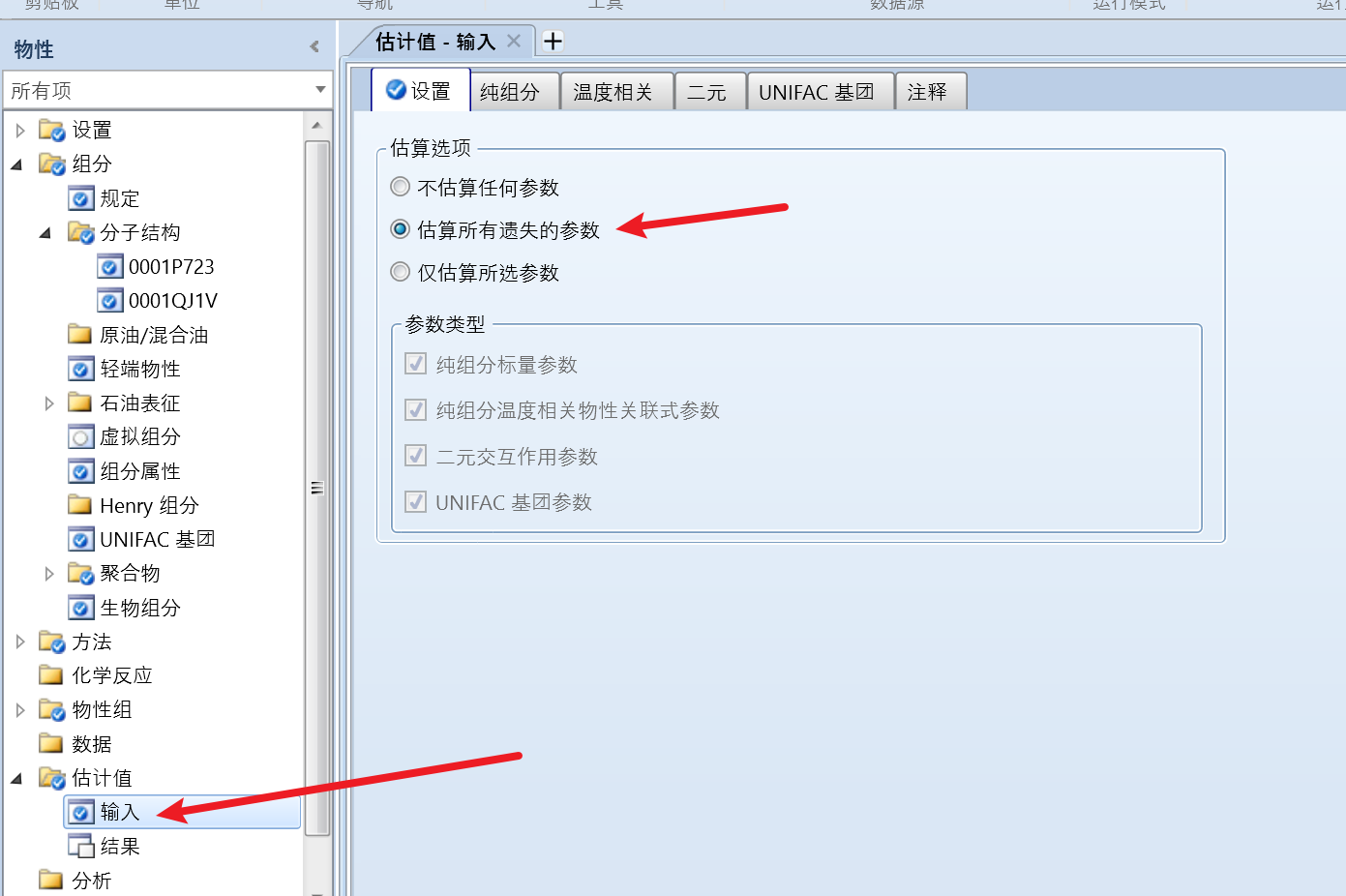
点击左侧估计值,输入,切换为估算所有遗失的参数:
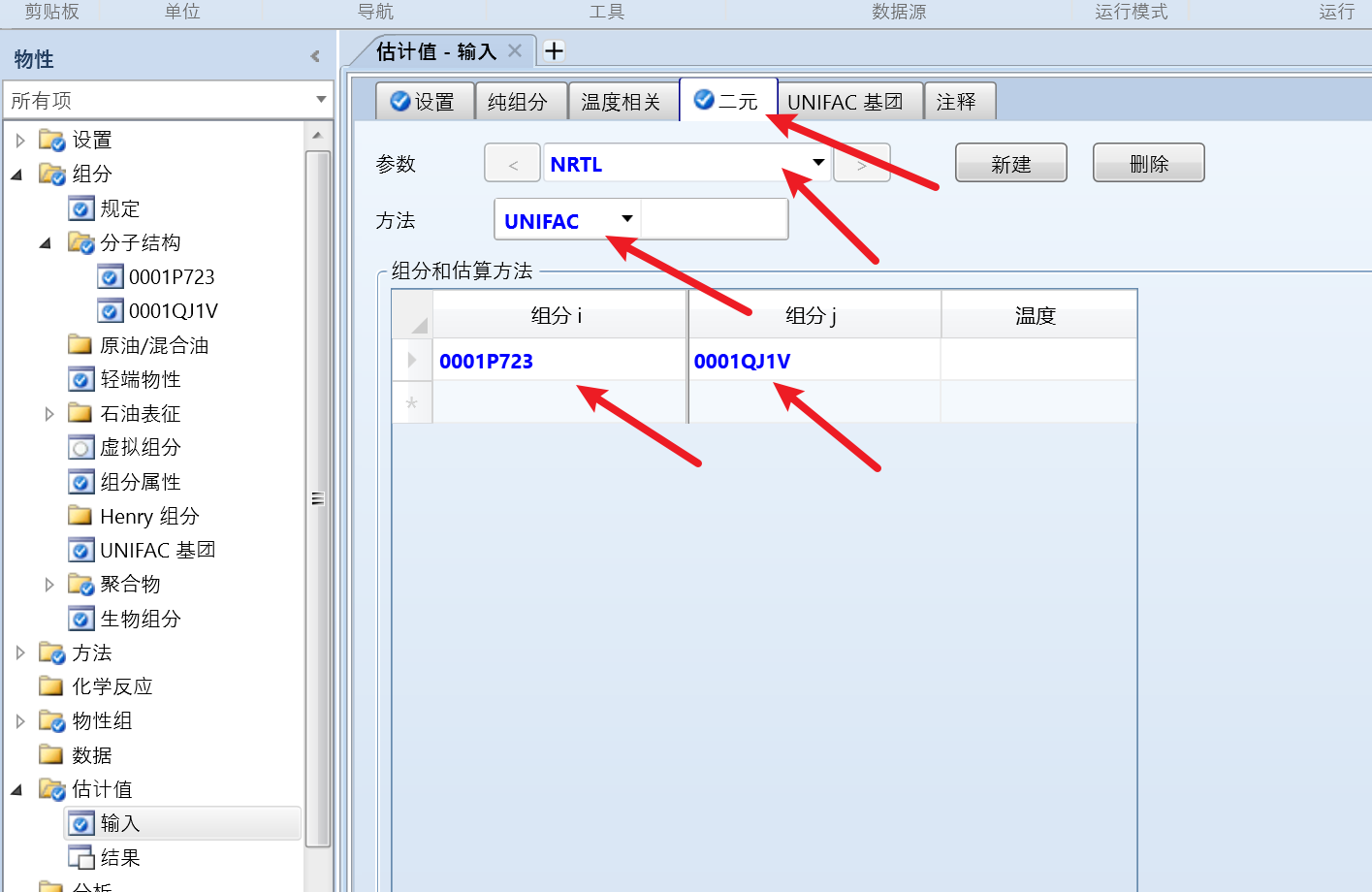
再点击二元选项卡,点击新建,选择物性方法和估算方法,选择组分:
输入估算温度区间,大概就是在两个物质的沸点区间即可:
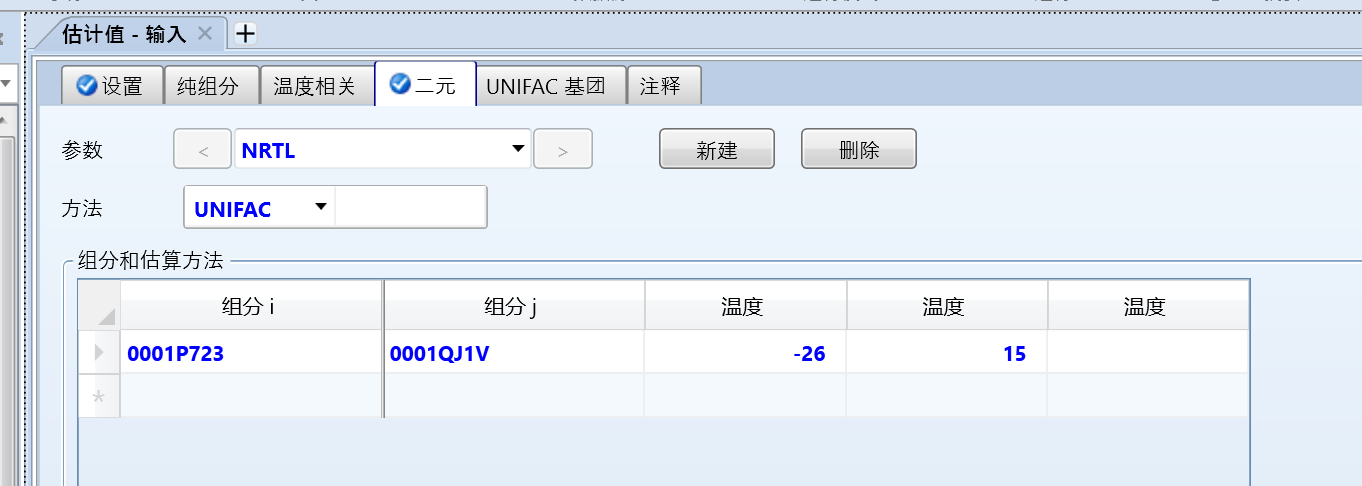
然后点击运行,即可估算出二者的交互参数:
这里需要补充一点,就是为什么我拿来做例子的这两个没有成功,反而一运行就警告,是因为分子结构的问题,导致键能计算的有误,
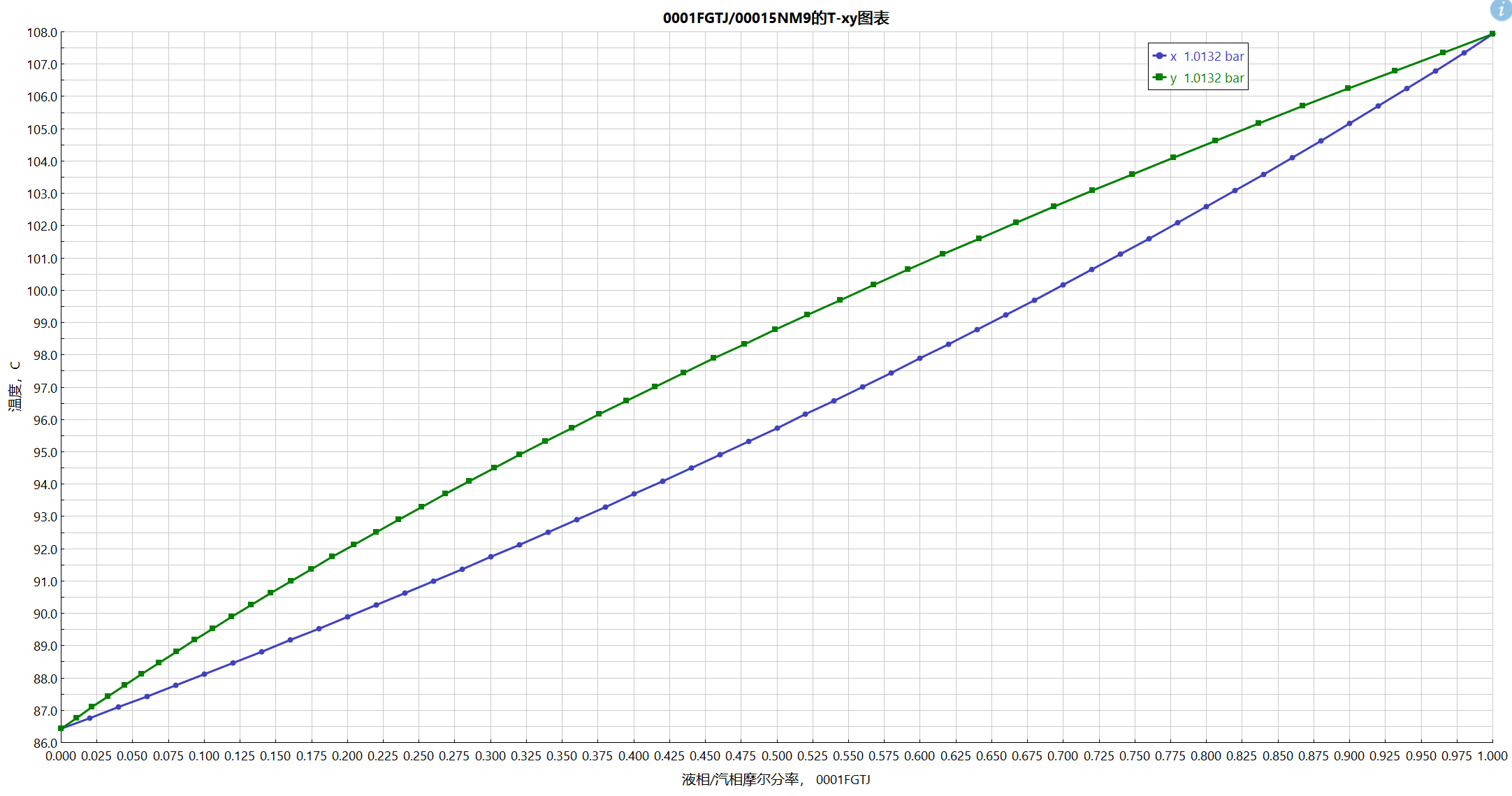
对比验证
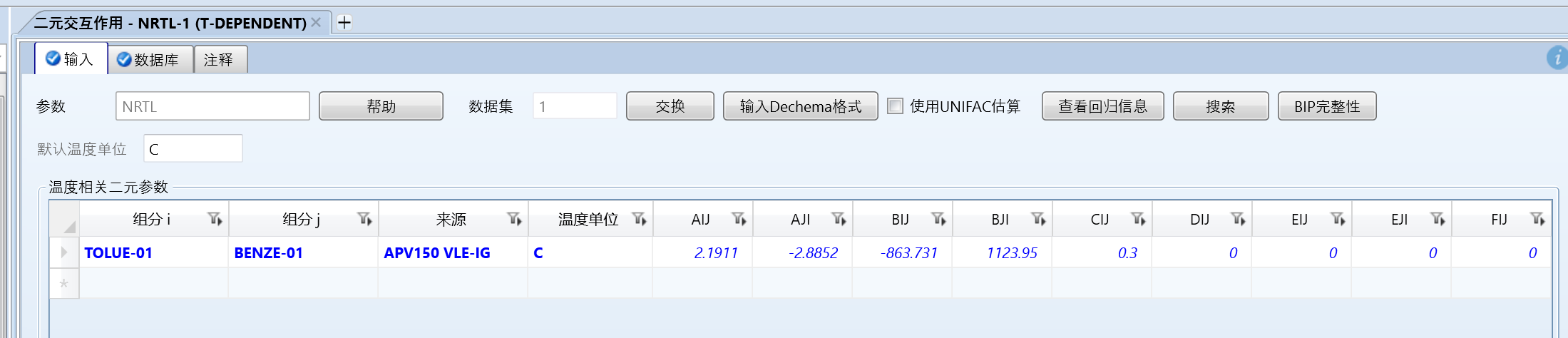
做一个简单的相图对比验证吧,以新的估算出来的苯和甲苯数据来做对比:
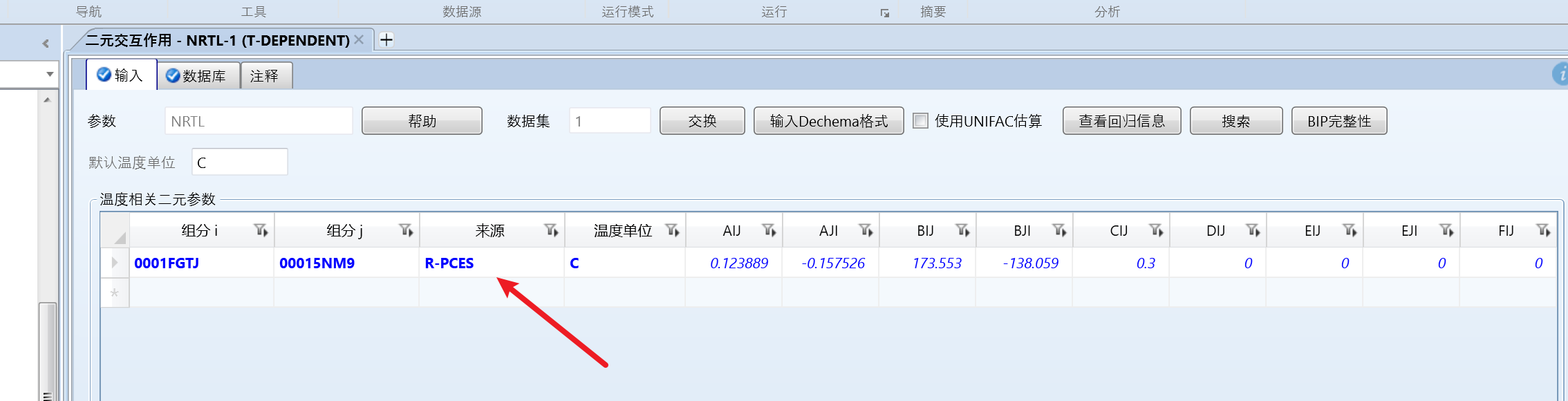
数据库中的数据:
估算数据:
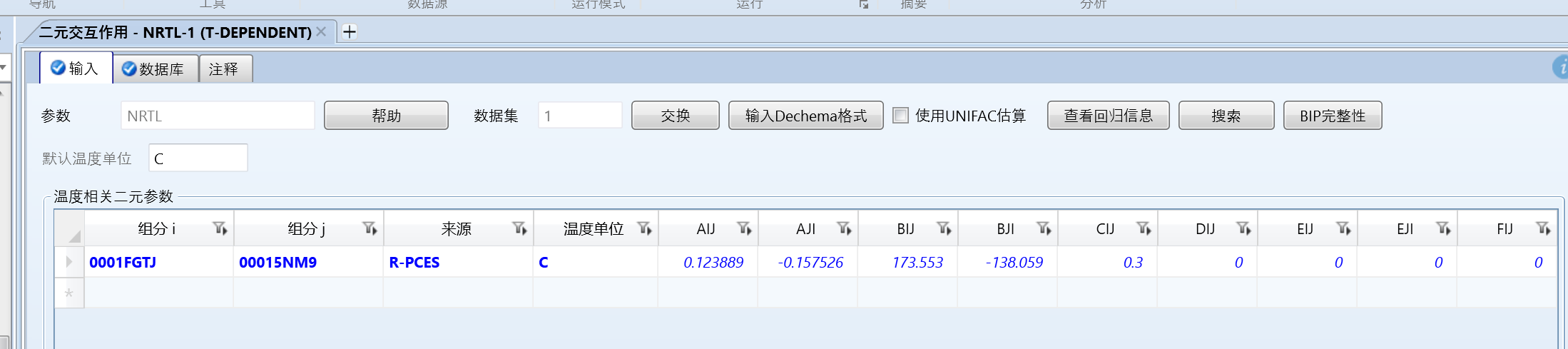
甲苯是0001-fgtj,苯是0001-5nm9,组分顺序反了。
数据库的相图:
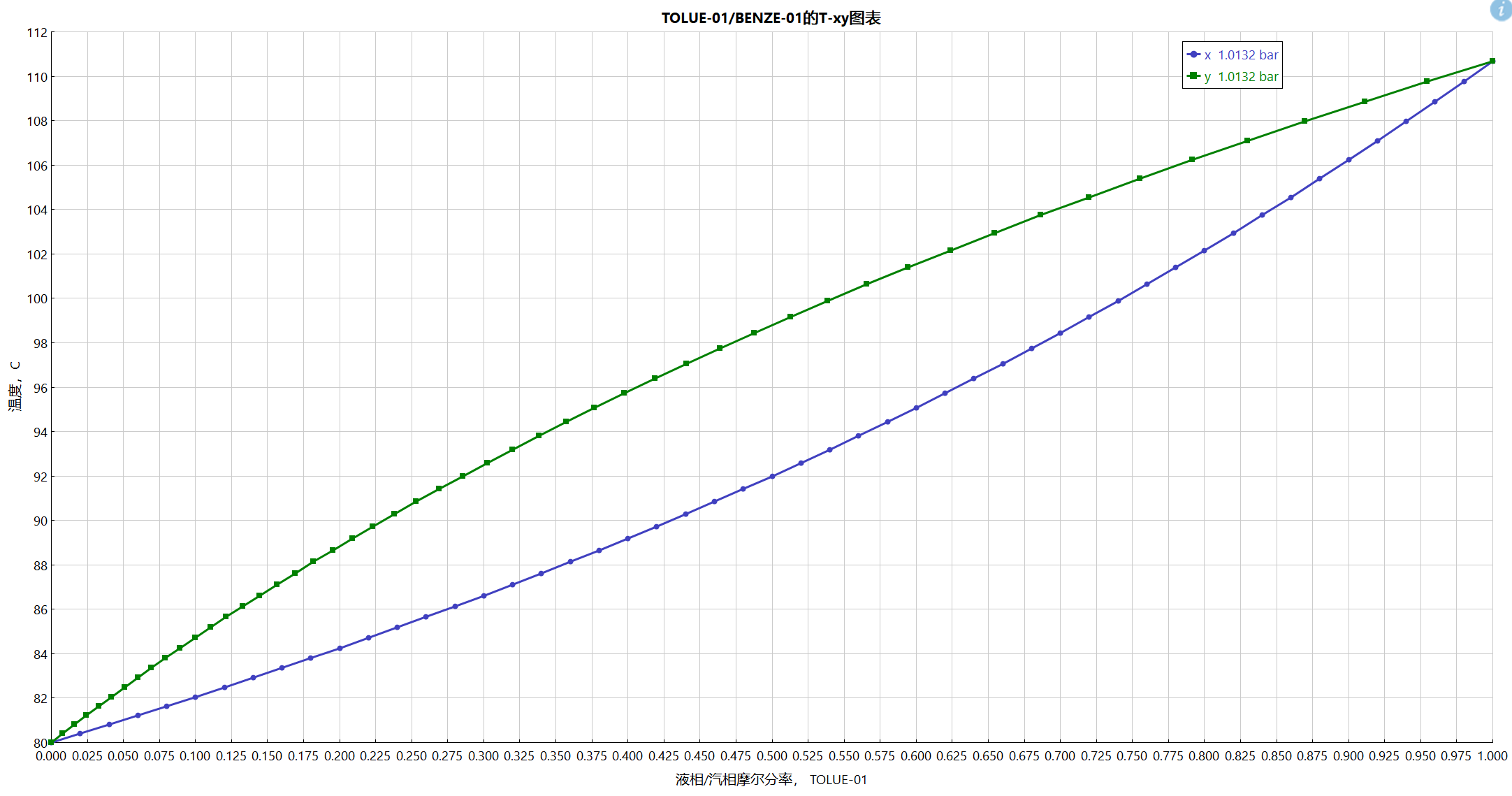
估算数据的相图: